
한국화학연구원에서 2021년 7월에 nature computational materials에 발표한 “Predicting thermoelectric properties from chemical formula with explicitly identifying dopant effects”를 읽고 내용을 공유합니다.
이전 포스팅도 참고 하시면 좋습니다.
Summary
- 머신 러닝은 소재 분야에서 많이 쓰이고 있습니다. 만들고자 하는 소재의 물성을 예측하고 어떻게 해야 원하는 특징을 가진 소재를 개발할 수 있을지 도움을 얻을 수 있기 때문입니다.
- 하지만 기존 방법들은 도핑된 물질에 대해선 정확한 특성 예측이 힘들어 전문가의 직관에 의존해 개발이 이루어졌습니다. 한국화학연구원은 이 문제를 해결하기 위한 알고리즘을 개발했고 DopNet이라 이름지었습니다.
- 기존 알고리즘의 문제는 크게 다음과 같습니다.
- 도핑된 재료의 결정 구조를 찾는데 너무 많은 계산이 필요하다.
- Dopants는 전체에서 낮은 비율을 차지하기 때문에 화학식을 기반으로 한 물질 표현은 해당 효과를 잘 나타내지 못한다(수치적 변화가 크지 않음).
- Dopants가 극적으로 host material의 성질을 바꿀 때 비선형이 크게 나타난다.
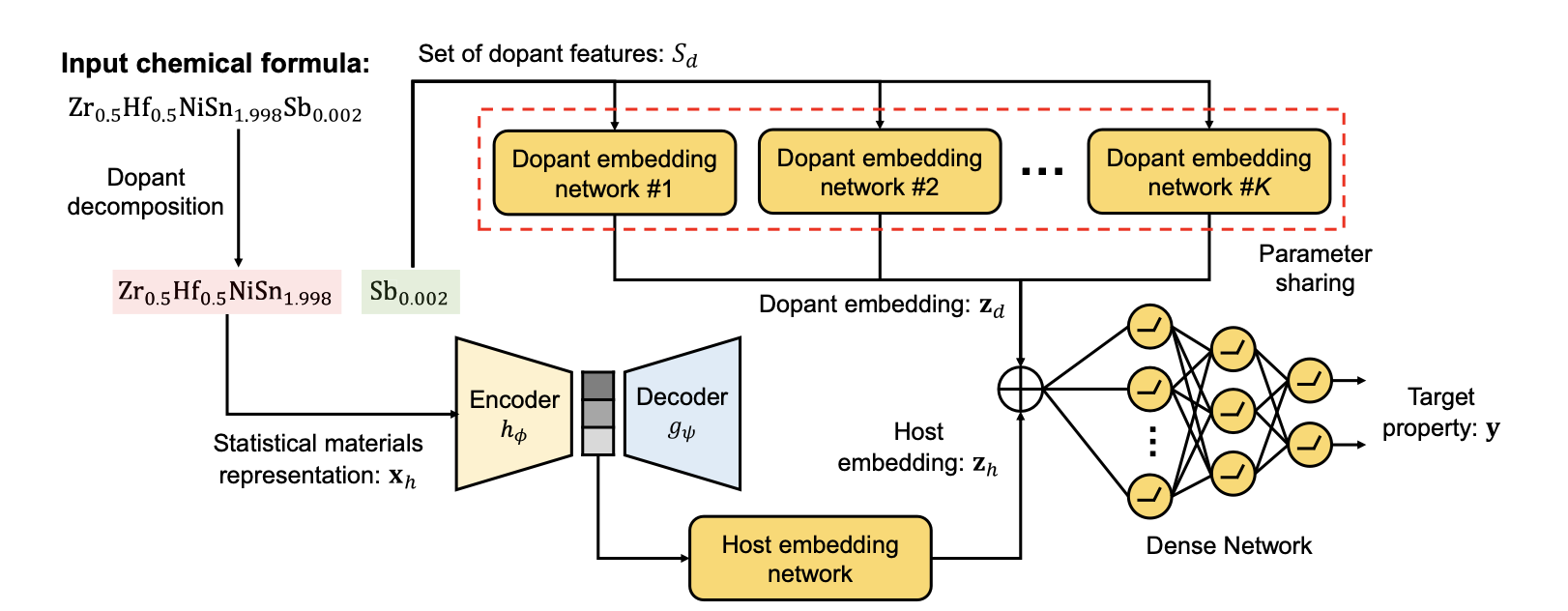
- 이를 해결하기 위해 DopNet은 결정 구조 계산 없이 재료 특성을 예측할 수 있도록 만들었습니다. 그리고 host material과 dopant를 명시적으로 나누어 임베딩하였습니다(기존 방법에서는 dopant 식별 여부와 관계 없이 재료 원자들에 대한 원소 통계 정보만 이용한 것으로 보입니다).
- DopNet은 세 부분으로 구성되어 있습니다.
- Host material 잠재 임베딩을 추출하는 호스트 임베딩 네트워크
- Dopants 잠재 임베딩을 생성하는 dopants 임베딩 네트워크
- Host material과 dopants의 임베딩을 통해 재료 특성을 예측하는 dense 네트워크
- DopNet이 작동하는 단계는 다음과 같습니다.
- 화학식이 입력되면 이는 host material과 dopants로 분해됩니다. 재료의 각 원자는 비율이 γ보다 작거나 같을 때 dopants로 분류되며, γ≥0은 DopNet에 미리 정의되어 있는 hyperparameter입니다.
- Host material는 원소 통계 정보를 기반으로 $\mathbf{x}_h$ 벡터로 표현됩니다. 호스트 특징 벡터에 인코더가 적용되고 호스트 임베딩 네트워크를 통과해 호스트 임베딩이 계산됩니다.
- Dopants의 특징 벡터는 최대 K개의 dopants를 포함할 수 있는 집합 $S_d$에 저장됩니다. 여기서 K는 hyperparameter입니다. Dopants는 dopant 임베딩 네트워크를 통해 임베딩되고 생성된 dopant 임베딩은 단일 벡터 $\mathbf{z}_d$로 표현됩니다.
- 마지막으로 dense network를 통해 물성 $\mathbf{y}$ 를 예측합니다.
Comments
- 이론 분야를 공부하고 있지만 실제 세상의 문제를 해결하기 위해 어떤 노력들과 아이디어도 흥미롭습니다. 특히 학부 전공과 관련된 내용들이다 보니 더 재미있게 읽었습니다.
- 내가 하는 연구나 공부가 실제로 어떻게 적용되어 어떤 문제를 해결하고 다른 이들의 연구와 인간의 삶에 어떤 영향을 미칠지 생각하거나 찾아 보는 것도 좋다고 생각합니다.